a. Sphingomyelinase
b. Hexosaminidase - A
c. Aryl sulphatase
d. Galactosidase - A
Niemann–Pick disease (/niːmənˈpɪk/ nee-mən-pik)[1] is a group of inherited severe metabolic disorders that allows a certain kind of fat to accumulate in cells. The fat, sphingomyelin, accumulates in lysosomes (membrane-bound organelles in cells). The lysosomes normally transport material through and out of the cell.


b. Hexosaminidase - A
c. Aryl sulphatase
d. Galactosidase - A
Niemann–Pick disease (/niːmənˈpɪk/ nee-mən-pik)[1] is a group of inherited severe metabolic disorders that allows a certain kind of fat to accumulate in cells. The fat, sphingomyelin, accumulates in lysosomes (membrane-bound organelles in cells). The lysosomes normally transport material through and out of the cell.
Signs and symptoms[edit]
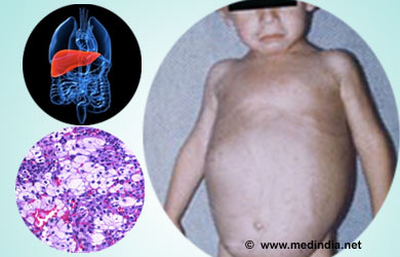
Symptoms are related to the organs in which sphingomyelin accumulates. Enlargement of the liver and spleen (hepatosplenomegaly) may cause reduced appetite, abdominal distension, and pain. Enlargement of the spleen (splenomegaly) may also cause low levels of platelets in the blood (thrombocytopenia)
Accumulation of sphingomyelin in the central nervous system (including the cerebellum) results in unsteady gait (ataxia), slurring of speech (dysarthria), and difficulty in swallowing (dysphagia).Basal ganglia dysfunction causes abnormal posturing of the limbs, trunk, and face (dystonia). Upper brainstem disease results in impaired voluntary rapid eye movements (supranuclear gaze palsy). More widespread disease involving the cerebral cortex and subcortical structures causes gradual loss of intellectual abilities, causing dementia and seizures.
Bones can also be affected: symptoms can include enlarged bone marrow cavities, thinned cortical bone, or a distortion of the hip bone called coxa vara. Sleep-related disorders, such as sleep inversion, sleepiness during the day and wakefulness at night, can occur. Gelastic cataplexy, the sudden loss of muscle tone when the patient laughs, is also seen.
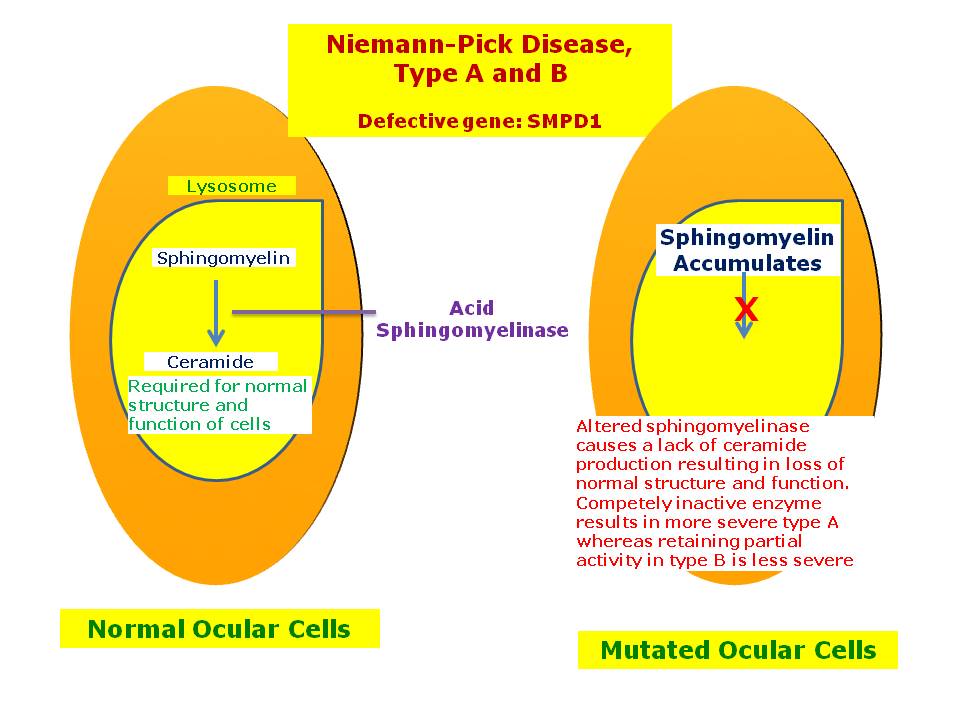
Mutations in the SMPD1 gene cause Niemann–Pick disease types A and B. They stop the body from making an enzyme, acid sphingomyelinase, that breaks down lipids.[3]
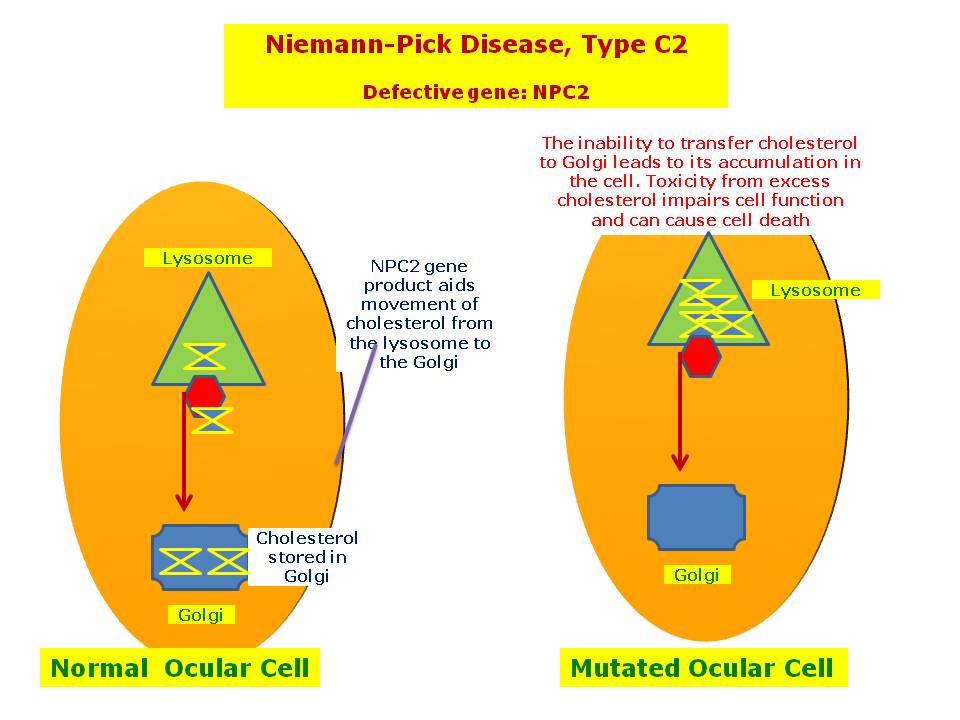
Mutations in NPC1 or NPC2 cause Niemann–Pick disease, type C (NPC), which affects a protein used to transport lipids.
Niemann–Pick disease is inherited in an autosomal recessive pattern, which means both copies, or alleles, of the gene must be defective to cause the disease.


Classification[edit]
- Niemann–Pick disease, SMPD1-associated, which includes types A and B
- Niemann–Pick disease type A: classic infantile
- Niemann–Pick disease type B: visceral
- Niemann–Pick disease, type C: subacute/juvenile, includes types C1 (95% of type C) and C2. Type C is the most common form of the disease[3] Type C2 is a rare form of the disease
Pathophysiology[edit]
Niemann–Pick diseases are a subgroup of lipid storage disorders called sphingolipidoses in which harmful quantities of fatty substances, or lipids, accumulate in the spleen, liver, lungs, bone marrow, and brain.
In the classic infantile type-A variant, a missense mutation causes complete deficiency of sphingomyelinase. Sphingomyelin is a component of cell membrane including the organellar membrane, so the enzyme deficiency blocks degradation of lipid, resulting in the accumulation of sphingomyelin within lysosomes in the macrophage-monocyte phagocyte lineage. Affected cells become enlarged, sometimes up to 90 μm in diameter, secondary to the distention of lysosomes with sphingomyelin and cholesterol. Histology shows lipid-laden macrophages in the marrow and "sea-blue histiocytes" on pathology. Numerous small vacuoles of relatively uniform size are created, giving the cytoplasm a foamy appearance
Ans. a. Sphingomyelinase
Pick's disease, a type of Fronto-temporal Dementia, is a rare neurodegenerative disease that causes progressive destruction of nerve cells in thebrain. Symptoms include dementia and loss of language (aphasia). While some of the symptoms can initially be alleviated, the disease progresses and patients often die within two to ten years.[1] A defining characteristic of the disease is build-up of tau proteins in neurons, accumulating into silver-staining, spherical aggregations known as "Pick bodies"
No comments:
Post a Comment